Heatmaps¶
bincs can be used to create heatmaps of ChIP-Seq data for predefined regions.
Example Output¶
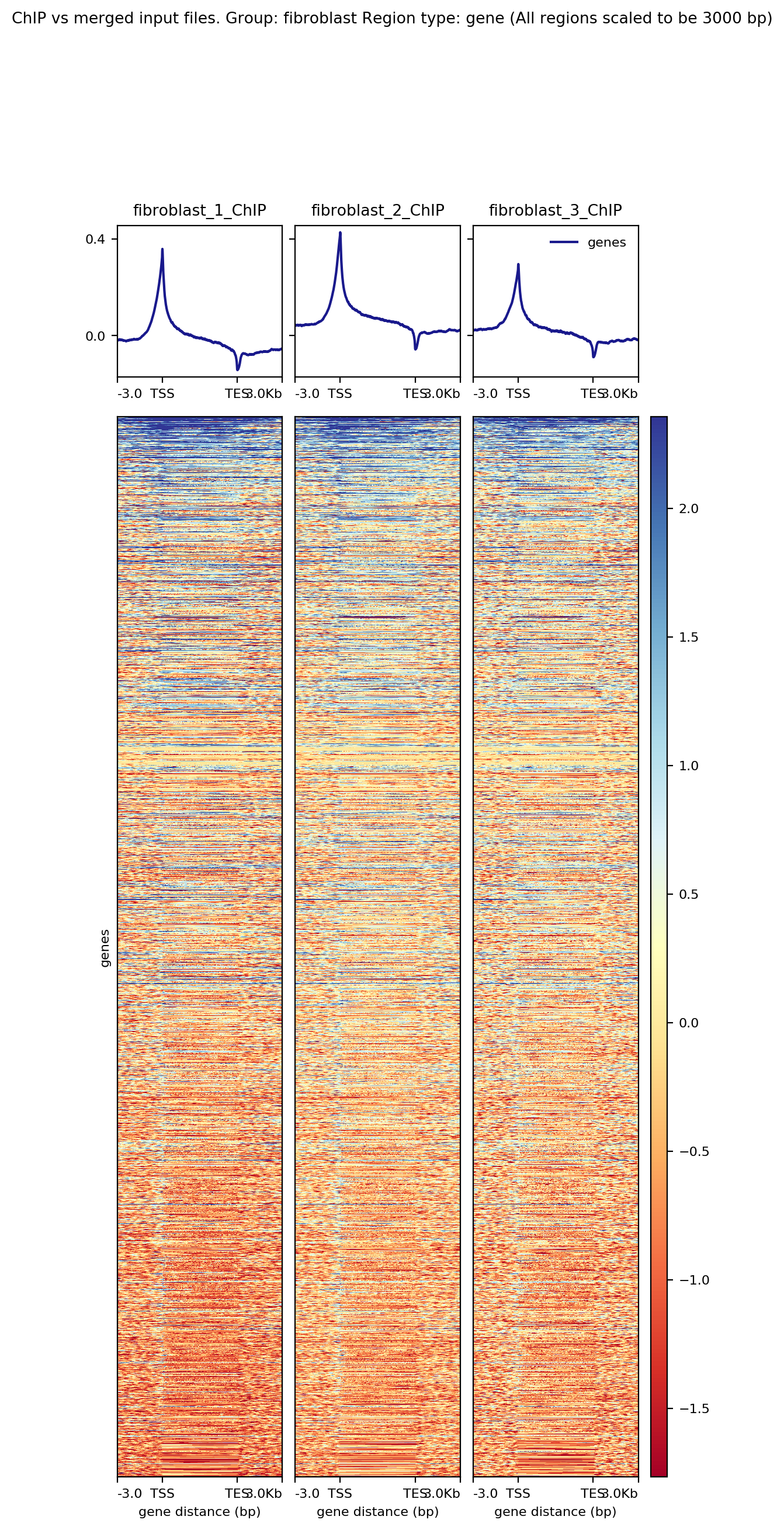
Fig. 1 A log2-ratio heatmap showing each sample against the pooled input for the group fibroblast.
Targets¶
There are three targets for creating regular heatmaps: log2_ratio_heatmaps, chip_heatmaps and input_heatmaps. The log2_ratio_heatmaps creates a heatmap of each ChIP sample against the pooled input for each group in the sample sheet. The chip_heatmaps and input_heatmaps create heatmaps of the RPKM-normalized ChIP or Input files for each group.
There is also a target for creating a heatmap of group vs group called log2_ratio_group_vs_group_heatmap. It creates one graph per group comparison of input-normalized ChIP groups against each other. This target requires that you have more than one group in your sample sheet.
Options¶
There are several settings that can be used to choose which regions should be included in the heatmaps and how much of each region to display.
Predefined Regions¶
The regions list in the config file can be used to select which regions should be graphed. If you are using a gencode gff3 The valid options are:
CDS, exon, five_prime_UTR, gene, start_codon, stop_codon,
stop_codon_redefined_as_selenocysteine, three_prime_UTR, transcript,
internal_exon
Using this list relies on having a gff3 annotation file set in your config. See https://www.gencodegenes.org/
If you do not have a gencode gff3, the UCSC refgene database will be used. Then the valid region options are:
regions:
- exon
- gene
- internal_exon
Custom Regions¶
If a you do not have a gff3 annotation available for your genome, you can use the custom_regions option in the config. This is a dict where the keys are region type names and the values are the path to a bed file denoting the regions.
custom_regions:
quantiles_internal_exon: test_data/WT.internal_exon.bed
quantiles_exon: test_data/WT.exon.bed
quantiles_gene: test_data/WT.gene.bed
The bed files should have a header and at least six columns. You can give an optional seventh column which must be called deepTools_group. It is used by deeptools to show each group separately in the profileplots. In the example below, the deepTools_group variable is used to show different quantiles of expression separately. The deepTools_group variable must be sorted. If a seventh column is used, you must use the exact header names shown below.
#chrom start end name score strand deepTools_group
chr2 241252956 241253035 exon:ENST00000391975.5:11 . - 0
chr9 133103746 133103833 exon:ENST00000424572.1:5 . - 0
chr17 32208106 32208309 exon:ENST00000584692.1:3 . + 0
chr17 32207511 32207563 exon:ENST00000584692.1:2 . + 0
chr17 82236728 82236872 exon:ENST00000584689.5:3 . + 0
chr17 82235982 82236231 exon:ENST00000584689.5:2 . + 0
chr9 133104262 133104331 exon:ENST00000424572.1:4 . - 0
chr9 133105931 133106016 exon:ENST00000424572.1:3 . - 0
chr9 133106644 133106748 exon:ENST00000424572.1:2 . - 0
...
chr5 122391088 122391191 exon:ENST00000509154.6:3 . + 75-100
chr5 122336787 122336904 exon:ENST00000509154.6:2 . + 75-100
chr1 160282038 160282200 exon:ENST00000392220.2:5 . - 75-100
chr1 160282416 160282502 exon:ENST00000392220.2:4 . - 75-100
chr1 160282943 160283109 exon:ENST00000392220.2:3 . - 75-100
chr1 160283529 160283639 exon:ENST00000392220.2:2 . - 75-100
chr12 98832028 98832136 exon:ENST00000552748.5:2 . - 75-100
chr12 98829173 98829353 exon:ENST00000552748.5:3 . - 75-100
chr4 59429 59556 exon:ENST00000509152.3:2 . + 75-100
chr1 36307769 36307825 exon:ENST00000505871.6:3 . + 75-100
Size of region around TSS/TES to graph¶
To set the size of the regions before the TSS and after the TSS to graph, use the flags
tss_distance_gene: 3000
tss_distance_other: 500
The setting tss_distance_gene will be used for all region names that contain “gene” in the name, otherwise the setting tss_distance_other will be used.
Sort order of heatmaps¶
To set the same sort order for all the heatmaps, you need to select one group to set as the default. All the heatmaps will then be sorted according to the sorting of this group.
sort_order_group:
If you want to have the group vs group heatmaps sorted (target log2_ratio_group_vs_group_heatmap) you need to provide a second sort order group:
second_sort_order_group:
Of course, the names of these groups must be the same as those used in the sample sheets.